Conceito de Southern Blotting: Uma Breve Introdução
O Southern Blotting ou Blot, foi batizado com o nome de seu inventor, Edward M. Southern que o desenvolveu para identificar sequências específicas de DNA. Pode ser usado para detectar a identidade, o tamanho e a abundância do DNA em uma mostra. Se um cientista quer saber se um organismo tem uma mutação genética particular, o Southern Blotting pode ser usado para identificar a presença desse gene mutante. A metodologia consiste na fragmentação do DNA em pedaços menores, através de uma eletroforese, seguindo-se a transferência para uma membrana e a detecção do fragmento de DNA de interesse por hibridação com uma sonda específica.
 |
As outras técnicas de transferência emergiram deste método e foram denominadas Northern Blotting (para RNA) e Western Blotting (para proteínas). |
Metodologia: Southern Blotting
Fragmentação
A primeira etapa é a fragmentação ou digestão do DNA, onde as longas sequências de nucleotídeos devem ser divididas em fragmentos menores com a ajudas das endonucleases ou enzimas de restrição. Cada enzima reconhece uma ou mais sequências alvo e corta o DNA nestas sequências ou perto delas. O corte é escalonado, produzindo terminações com DNA fita simples.
|
Uma das primeiras enzimas de restrição a ser isolada foi a EcoRI, foi produzida pela bactéria Escherichia coli. Essa enzima reconhece apenas a sequência GAATTC |
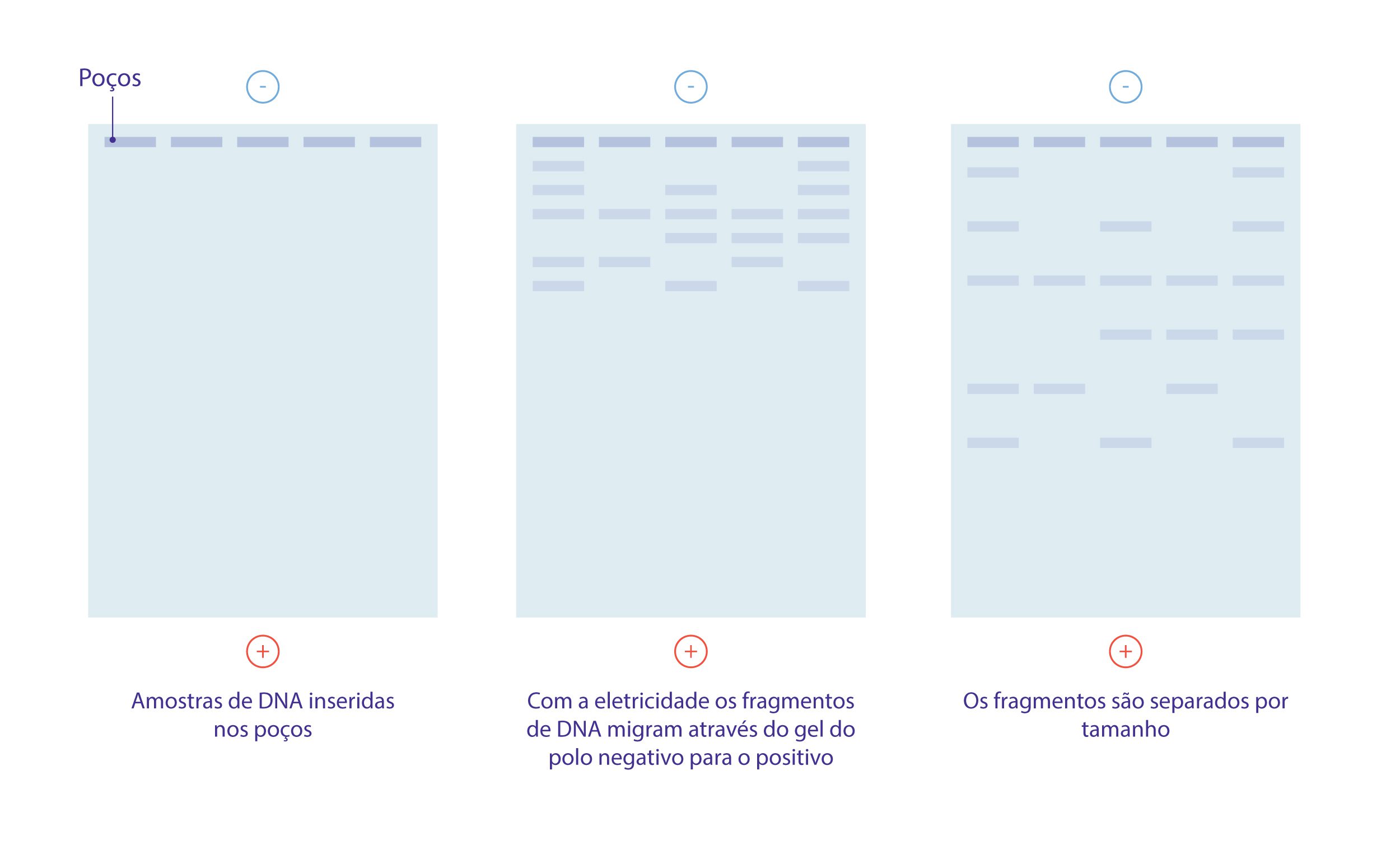
Eletroforese em gel de agarose
O DNA fragmentado é submetido a eletroforese em gel de agarose para separar os fragmentos de acordo com peso molecular à medida que passam por uma corrente elétrica.
Todas as moléculas de DNA têm a mesma quantidade de carga por massa. Por essa razão, a eletroforese em gel de fragmentos de DNA faz a separação baseada apenas no tamanho. Através do uso da eletroforese, podemos ver quantos diferentes fragmentos de DNA estão presentes em uma amostra, e o quão grande eles são em relação aos outros. Podemos também determinar o tamanho absoluto de um pedaço de DNA examinando-o ao lado de um DNA “padrão”, composto de fragmentos de DNA de tamanhos conhecidos.
Transferência do DNA para uma membrana
Para transferência do DNA separado no gel para uma membrana de nitrocelulose ou de nylon, é necessário colocar a membrana sobre o gel e aplicar pressão uniformemente distribuída sobre o gel, levando à ligação do DNA à membrana. Seguidamente, a membrana é aquecida (no caso da utilização de uma membrana de nitrocelulose) ou exposta a radiação UV (no caso da utilização de uma membrana de nylon) para fixar permanentemente o DNA à membrana.
Recomenda-se a utilização de membranas de nylon devido à sua capacidade de se ligar mais eficientemente ao DNA.
Marcação da sonda
A sonda é um pequeno pedaço de DNA que é projetada para ter uma sequência complementar a sequência particular do DNA na amostra. Isto permite que a sonda hibride ou se ligue a um fragmento de DNA específico na membrana. Além disso, a sonda possui um marcador, que normalmente é um átomo radioativo ou um corante fluorescente.
Hibridação
A técnica de hibridação é importante para analisar genes expressos assim como genes não expressos na amostra. A visualização destes genes de interesse dá-se pelo emparelhamento das sondas que se encontram marcadas.
Assim, após a hibridação, a sonda permite que o fragmento de DNA de interesse seja detectado entre os muitos fragmentos de DNA diferentes na membrana. Essa sonda irá parear com qualquer sequência de DNA complementar a ela. Portanto se a sequência procurada não estiver presente na amostra não houve o pareamento.
Lavagem e Detecção
O excesso de sonda é lavado da membrana, e toda a sonda não hibridizada será removida. Depois disso, a presença ou não da sonda na membrana (e as posições onde ela está presente) é revelada por uma autorradiografia em um filme de raio-X, ou pelo aparecimento de uma cor caso um marcador cromogênico tenha sido usado.
Aplicação
- Detectar DNA em determinada amostra
- Testes de paternidade, identificação criminal, identificação de vítimas
- Para isolar e identificar um gene de interesse
- Usado no polimorfismo de comprimento de fragmento de restrição
- Identificar mutação ou rearranjo gênico na sequência do DNA
- No diagnóstico de doenças causadas por defeitos genéticos
- Identificar agentes infecciosos
Introdução do PCR em Tempo Real
Assim como computadores, telefones celulares e carros se tornam tecnologicamente mais avançados, deixando as versões anteriores obsoletas, algumas técnicas laboratoriais são substituídas por versões aprimoradas que economizam tempo e dinheiro. No entanto, as técnicas tradicionais e até mesmo históricas, não deixam de perder sua importância no campo científico.
Hoje, o Southern blotting pode ser substituído pelo PCR em tempo real para responder às mesmas questões experimentais. O PCR em tempo real detecta e quantifica um gene ou sequência de DNA de interesse registrando a abundância de DNA ao longo do processo de amplificação. Isso permite ao pesquisador comparar diferentes sequências de DNA para ver qual é mais abundante em uma amostra.
O Southern blotting é intensivo em mão-de-obra e requer uma grande quantidade de DNA de alta qualidade, enquanto que no PCR em tempo real a automação é mais fácil, a quantidade de DNA necessário é bem menor, além da economia do tempo e recursos. Porém, e metodologia Southern blotting a taxa de falsos positivos é mais baixa do que o PCR em tempo real.
Produtos Kasvi




Referências:
- https://www.mybiosource.com/learn/southern-blotting/
- https://www.thermofisher.com/br/en/home/life-science/dna-rna-purification-analysis/nucleic-acid-gel-electrophoresis/southern-blotting.html
- http://www.onlinebiologynotes.com/southern-blotting-principle-procedure-application/
- https://www.news-medical.net/life-sciences/What-is-Southern-Blotting.aspx
- https://blog.addgene.org/evolution-of-lab-techniques
- https://www.sobiologia.com.br/conteudos/Biotecnologia/enzimasderestricao.php
- https://pt.khanacademy.org/science/biology/biotech-dna-technology/dna-sequencing-pcr-electrophoresis/a/gel-electrophoresis
Compartilhe nas redes sociais:
Lançamentos

















Artigos relacionados